
Gentherapie
Auch im Zeitalter moderner Medizin sind viele schwere Erkrankungen nach wie vor nicht behandelbar. Viele dieser Leiden sind genetisch bedingt. Seit einigen Jahren macht die Gentherapie als neue Ära der Medizin von sich reden. Doch ist sie wirklich in der Lage, Krankheiten durch einfache Reparatur unseres Erbgutes dauerhaft zu heilen? Wo liegen Chancen, wo Herausforderungen, wo Risiken? Wir beleuchten den Stand der Technik in Deutschland und Europa.
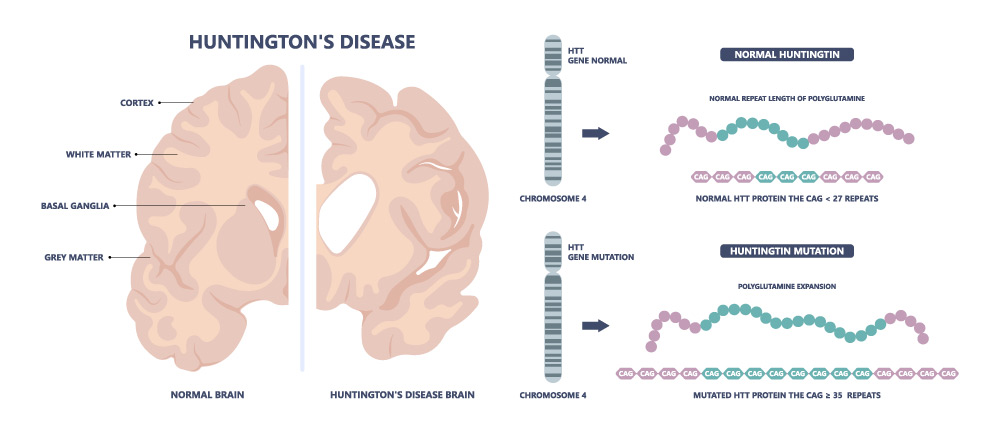
Rund 3 100 000 000 (3,1 x 1010) Basenpaare besitzt das menschliche Genom. Schon drei Basenpaare mehr können über Leben und Tod entscheiden. CAG, kurz für die drei DNA-Basen Cytosin, Adenin und Guanin, lautet die fatale Abfolge bei der erblichen, stets tödlich verlaufenden Nervenkrankheit Chorea Huntington, dem ersten Erbleiden, für das Forschende vor rund 30 Jahren das verantwortliche Huntington-Gen auf dem menschlichen Chromosom 4 fanden. Im genetischen Code des gesunden Gens findet man die Abfolge CAG in 10 bis 26-facher Wiederholung. Das Basentriplett codiert für die Aminosäure Glutamin, das resultierende Huntington-Protein, kurz HTT, enthält also auch bei Gesunden einen Polyglutaminbereich. Das krankhafte Huntington-Gen indes wächst im Verlauf der Zellteilungen und immer mehr CAG-Abfolgen kommen hinzu – ein verändertes HTT entsteht. Ab 40 Wiederholungen gilt es als sicher, dass die betroffene Person erkranken wird, je mehr CAG-Einheiten direkt aufeinander folgen, desto früher bricht die Krankheit aus. Aber: Ist die Kette der CAGs beispielsweise durch nur eine einzige CAA-Sequenz unterbrochen, bleiben die Betroffenen länger verschont.
Über kaum eine andere Erberkrankung ist so viel bekannt wie über die als „Seltene Erkrankung“ geltende Chorea Huntington. Das fatale Leiden dient mittlerweile als Modellerkrankung, an der Forschende für andere Erbkrankheiten lernen – heilen lässt es sich mangels einer kausalen Therapie bisher jedoch nicht. Doch müsste sich der ursächliche Defekt am Erbgut im Zeitalter von Hightech-Medizin, Molekulargenetik und Gentherapie nicht „reparieren“ lassen? Was versteht man eigentlich unter Gentherapie und sind wir überhaupt in ihrem Zeitalter angekommen? Für die Behandlung welcher Erkrankungen wird die Gentherapie in Deutschland bereits eingesetzt und wo liegen wesentliche Herausforderungen? Und kann eine Gentherapie Erbkrankheiten wirklich ursächlich heilen, statt nur Symptome zu behandeln?
Als Gentherapeutikum bezeichnet man per Definition des Paul-Ehrlich-Instituts „ein biologisches Arzneimittel, dessen Wirkstoff eine Nukleinsäure (Träger der Erbinformationen) enthält oder daraus besteht. Es wird eingesetzt, um eine Nukleinsäuresequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu entfernen. Die therapeutische, prophylaktische oder diagnostische Wirkung steht in unmittelbarem Zusammenhang mit der rekombinanten Nukleinsäuresequenz, die es enthält oder mit dem Produkt, das auf Basis dieser genetischen Information gebildet wird.“
Gentherapeutika zählen laut einer Ausarbeitung des Deutschen Bundestages von 2024 „zu den so genannten Arzneimitteln für neuartige Therapien, kurz ATMP (Advanced Therapy Medicinal Products) gemäß der Arzneimittel-Richtlinie 2001/83/EG1 und der ATMP-Verordnung (EG) 1394/20072. ATMP sind in der EU wie Arzneimittel und darüberhinausgehend entsprechend der genannten ATMP-Verordnung reguliert. […] Für ihre Marktzulassung müssen ATMP wie jedes andere Arzneimittel das zentralisierte Zulassungsverfahren der EU bei der Europäischen Arzneimittel-Agentur (EMA) durchlaufen.“
Genetherapie – auf Euphorie folgte Ernüchterung
Den Anfang dieser völlig neuen Ära der Medizingeschichte markiert indes bereits der 14. September 1990. An diesem Tag behandelten die US-amerikanischen Ärzte French Anderson und Michael Blaese die damals vierjährige Ashanti De Silva im Rahmen einer klinischen Studie erstmals mit einer retroviralen Gentherapie. Das Mädchen, das damit zur weltweit ersten gentherapeutisch behandelten Patientin wird, leidet an der genetisch bedingten Immunschwäche ADA-SCID: Ihr fehlt ein Enzym, das die für die Immunabwehr so wichtigen T-Lymphozyten vor dem Angriff eines anderen Proteins schützt. In der Folge reifen die T-Lymphozyten in ihrem Knochenmark nicht oder nur in zu geringer Zahl heran, wodurch für sie alle Krankheitserreger eine potenziell tödliche Gefahr sind. Die meisten Patienten mit dieser Erkrankung überleben das Kindesalter nicht. French Anderson und Michael Blaese entnahmen dem Mädchen einige ihrer verbliebenen weißen Blutkörperchen und gaben im Labor ein zuvor genetisch verändertes und mit einer funktionierenden Kopie des defekten Gens ausgestattetes Retrovirus hinzu. Dieses fungiert als „Genfähre“ und schleust das korrekte Gen in die Immunzellen des Mädchens ein. Die auf diese Weise veränderten Zellen wurden ihr anschließend wieder per Infusion verabreicht. Heute bestehen Zweifel daran, ob nicht ein zweites Medikament, welches Ashanti verabreicht wurde, zum Erfolg beitrug – doch das Mädchen ist heute eine junge Frau und lebt.
Ashantis Fall löste unter Forschern und Medizinern Euphorie aus. Viele sahen eben jenes neue Zeitalter eingeläutet, in dem schwerste Erberkrankungen oder andere Leiden mit genetischer Beteiligung wie Krebs, Immun- und Stoffwechselkrankheiten heilbar sein würden. Doch auf die Euphorie folgte Ernüchterung. Ausgerechnet die als „Genfähren“ verwendeten viralen Vektoren führten bei einigen Patienten zu schwersten Nebenwirkungen und Komplikationen. Bei manchen kam es zu schweren überschießenden, sogar tödlichen Immunreaktionen, andere entwickelten in der Folge der Behandlung beispielsweise Leukämien. Die „Gentaxis“ waren offenbar zu unsicher und unspezifisch.
Sichere „Fähren“ für Gentherapeutika in Arbeit
Viren sind als „Genfähren“ zunächst einmal prädestiniert. Denn was sie richtig gut können, ist ihr Genmaterial in andere Wirtszellen einzuschleusen und dort mithilfe der wirtszelleigenen Maschinerie replizieren zu lassen. Nur so können sie sich vermehren.
Aber keines der zwei aktuell verwendeten Systeme funktioniert perfekt: Die häufig verwendeten Adenoviren bringen das Genmaterial zwar hoch effizient, aber nur vorübergehend, in die Zelle ein, weshalb eine auf solchen Vektoren basierende gentherapeutische Behandlung meist zeitlebens regelmäßig wiederholt werden muss. Außerdem senken diese Vektoren zwar das Risiko von „Fehleinbauten“ in das Genom der Patientinnen und Patienten, lösen aber als (abgewandelte) Erreger von beispielsweise grippalen Infekten oft heftige Immunreaktionen aus. Retroviren hingegen schleusen das therapeutische Gen dauerhaft in das Genom ein – im Idealfall kann auf diese Weise eine einzige Injektion des Gentherapeutikums ausreichen, um den Gendefekt zu beheben und die Erkrankung zu heilen. Allerdings erfolgt der Einbau des Genmaterials mitunter mehrfach und unspezifisch auch an anderer Stelle im Genom, was letztlich auch Krebs auslösen kann.
Neue Wege und Vektoren, die genetische Information sicher, effizient und gezielt in die Zellen der Patientinnen und Patienten einschleusen zu können, waren demnach dringend gesucht und sind bis heute Gegenstand intensiver Forschung. Insbesondere Fortschritte auf dem Gebiet der Gentechnik, wie zuletzt die CRIPRS/Cas9-Technik, schufen die Möglichkeit, Vektoren immer gezielter zu designen. Auf diese Weise ließen sich insbesondere in den letzten zwei Jahrzehnten die Sicherheit und Spezifität einiger viraler Vektoren immer weiter erhöhen, was ihnen zu einer Art Renaissance verhalf.
Ein Ansatz bestand dabei darin, diese Vektoren immer weiter auf die gerade noch für den effizienten Gentransfer notwendigen Funktionen zu verkleinern und gleichzeitig die Spezifität des Einbaus zu erhöhen. Das erste Gentherapeutikum, das am 2. November 2012 die offizielle Marktzulassung für Europa erhielt, basiert auf einem entkernten Minivirus, dem so genannten Adeno-assoziierten Virus (AAV). Diese extrem kleinen Viren hinterlassen ihr Erbgut (mit dem Gentherapeutikum) als extrachromosomales DNA-Stück in den Zellkernen der Zielzellen, wo es von der zelleigenen Maschinerie abgelesen wird. In diesem Fall sollte das von der in Amsterdam ansässigen Firma UniQure entwickelte Präparat „Glybera“ Patientinnen und Patienten helfen, die an der erblichen Stoffwechselkrankheit Lipoprotein-Lipase-Defizienz (LPLD) leiden. Die Erkrankung lässt bestimmte Blutfette der Betroffenen krankhaft ansteigen. Aus wirtschaftlichen Gründen wurde das Medikament allerdings wieder vom Markt genommen.
Gentherapie – State of the art in Deutschland
Grundsätzlich lassen sich zwei verschiedene Ansätze der Gentherapie unterscheiden.
Die Modifikation des Erbguts von Körperzellen (somatischen Zellen), beschränkt sich auf die individuellen Patientinnen und Patienten (somatische Gentherapie). Eine Weitergabe der therapierten Gensequenz an deren Nachkommen erfolgt nicht. Wird hingegen in das Erbgut von Keimzellen (Spermien oder Eizellen oder deren Vorstufen) eingegriffen, wird nicht nur das Erbgut der einzelnen Patientinnen und Patienten verändert, sondern auch das der Nachkommen (Keimbahntherapie). Da die Keimbahntherapie ein höheres Risiko unvorhersehbarer Konsequenzen birgt, erfordert sie umfassendere technische und ethische Bewertungen. In Deutschland ist sie gemäß § 5 des Embryonenschutzgesetzes (ESchG) verboten. Alle Gentherapien, die hier bisher entwickelt und in klinischen Studien geprüft wurden, sind somatische Gentherapien.
Hinsichtlich des Einbringens des gewünschten therapeutischen Gens bzw. des Werkzeugs zur Reparatur von Genen in die richtigen Zellen unterscheidet man in vivo-Ansätze, bei denen das Therapeutikum direkt verabreicht und die Zielzellen direkt im Organismus anvisiert werden, von so genannten ex vivo-Ansätzen. Hierbei werden den Zielpersonen Zellen, beispielsweise Stammzellen aus dem Knochenmark, entnommen, im Labor genetisch verändert und anschließend wieder verabreicht. Häufig werden darüber hinaus noch in situ-Ansätze unterschieden, bei denen das Therapeutikum direkt in das betroffene Organ bzw. eine spezifische Stelle eingebracht wird.
Die ex vivo-Strategie wird auch als zellbasierte Gentherapie bezeichnet und findet insbesondere bei der Krebstherapie Anwendung. Im Fall der so genannten CAR-T-Zell-Therapie werden den Patientinnen und Patienten T-Lymphozyten entnommen und genetisch so modifiziert, dass sie Krebszellen über spezifische Antigenrezeptoren auf deren Oberfläche erkennen und angreifen können. Bemerkenswert an dieser sogenannten N=1-Therapie ist, dass das Therapeutikum auf eine einzelne Zielperson (Anzahl „N“ = 1) zugeschnitten wird und somit die optimale Wirkung entfalten kann. Diese so genannte Krebsimmuntherapie gilt als äußerst vielversprechend für die Behandlung bestimmter Krebsarten. In Deutschland sind, Stand heute, sechs der insgesamt 16 zugelassenen Gentherapeutika CAR-T-Zell-Therapien (siehe Tabelle). Auf diesem Gebiet wird intensiv geforscht, weshalb in Zukunft wohl von weiteren Zulassungen auszugehen ist. Neben malignen Erkrankungen, wie Leukämien und Lymphome, stellen angeborene Erkrankungen des Blutgerinnungssystems, wie Hämophilie A und B, ein weiteres Hauptanwendungsgebiet der derzeit in Deutschland zugelassenen Gentherapeutika dar.
Die überwiegende Mehrzahl aller hierzulande zugelassenen Gentherapeutika richtet sich gegen so genannte „Seltene Erkrankungen“. Da diese zu etwa 80 Prozent genetisch bedingt oder mitbedingt sind, wird der Gentherapie großes Potenzial für eine verbesserte Versorgung von Betroffenen zugeschrieben. Insbesondere monogenetisch bedingte Erkrankungen, also solche, für die nur ein einzelnes Gen ursächlich verantwortlich ist, gelten als gentherapeutisch adressierbar und werden zum Teil intensiv beforscht.
Als „selten“ gilt eine Krankheit hierzulande, wenn nicht mehr als einer von 2000 EU-Bürgern an ihr leidet. Für die zur Behandlung dieser Erkrankungen entwickelten Wirkstoffe, können Unternehmen unter anderem einen so genannten Orphan-Drug-Status beantragen. In der Arzneimittelzulassung führt dieser zwar nicht zu einem beschleunigten Verfahren, dennoch erhalten Zulassungsinhaber einige Vorteile wie beispielsweise eine zehnjährige Marktexklusivität innerhalb der EU.
Anlass zur Hoffnung auch bei Chorea Huntington?
Und Chorea Huntington? Können davon Betroffene in Zukunft ebenfalls auf eine gentherapeutische Behandlungsmöglichkeit hoffen? Auch diese Seltene Erkrankung hat eine monogenetische Ursache und ist noch dazu gut erforscht. Tatsächlich arbeiten derzeit viele Unternehmen und Forschende an möglichen Gentherapien, um das betroffene Huntington-Gen zu korrigieren oder „auszuschalten“.
Der derzeit aussichtsreichste Wirkstoffkandidat ist wohl AMT-130 der Firma uniQure – der Firma also, die 2012 die erste Marktzulassung für ein Gentherapeutikum in Europa erhielt. Im Sommer letzten Jahres verkündete das Unternehmen in einer Halbzeitbilanz, dass ihre Gentherapie in zwei klinischen Phase-I/II-Studien nach zwei Jahren Beobachtungszeit den Niedergang der Probanden klar verzögert: in einer niedrigen Dosierung um 30 Prozent, in einer höheren jedoch um ganze 80 Prozent, was nahezu einem Stillstand der Erkrankung gleichkommt. In der „Huntington-Szene“ löste diese Nachricht vorsichtige Hochstimmung aus – wenn ein Effekt so deutlich von der Dosis eines Wirkstoffs abhängt, gilt das in der Pharmaforschung und -entwicklung als gutes Zeichen dafür, dass die gemessene Wirkung real ist.
Der Gentherapie-Produktkandidat AMT-130 besteht nach Angaben des Unternehmens aus einem AAV5-Vektor (Adeno-assoziierter Virus Typ 5), der eine künstliche Mikro-RNA trägt, die die Ablesung des Huntington-Gens in den Zellen der Patientinnen und Patienten durch einen nicht-selektiven Knockdown unterbindet. Therapeutisches Ziel ist demnach, die Produktion des mutierten, für die Nervenzellen der Betroffenen toxischen, Proteins mHTT zu hemmen. Auch die US-amerikanische Arzneimittelbehörde FDA hält das Mittel offenbar für so aussichtsreich, dass sie ihm als ersten therapeutische Kandidaten den RMAT-Status für die Huntington-Krankheit erteilte (Regenerative Medicine Advanced Therapy). Der RMAT-Status ermöglicht eine verstärkte Zusammenarbeit mit der FDA, um die Entwicklung zu beschleunigen und möglicherweise eine schnellere Zulassung zu erreichen. Auch der Orphan-Drug-Status wurde dem Mittel bereits erteilt.
Die Chorea-Huntington-Therapie wird vor allem dadurch erschwert, dass sich die Krankheit in bestimmten Zellen des Gehirns der Betroffenen abspielt und das Medikament auch genau dort hingelangen muss. Ein Medikament jedoch ins Gehirn zu bringen und somit die so genannte Blut-Hirn-Schranke zu überwinden ist alles andere als trivial – eine Genkorrektur in den Nervenzellen des Gehirns noch ungleich schwieriger. Aktuell muss AMT-130 den Therapierten daher direkt ins Hirn gespritzt werden, was sie im Angesicht der Ausweglosigkeit dieser Erkrankung in Kauf nehmen.
Die Liste schwerster Erkrankungen, die nach wie vor kaum therapierbar sind, ist indes lang. Allein etwa 8000 Krankheiten weltweit, zählen zu den Seltenen Erkrankungen, die in der Medizin aus unterschiedlichen Gründen ohnehin unterrepräsentiert sind. In vielen Fällen entsteht enormes Leid, weder Krankheit noch wie auch immer geartete Therapie sind ein „Spaziergang“. Besteht eine berechtigte Chance auf Linderung oder Heilung, kann die Gentherapie eine ganz individuelle Hoffnung und Entscheidung sein.
In Deutschland zugelassene Gentherapeutika (Stand: 14.03.2025; Quelle: Paul-Ehrlich-Institut, Gentherapeutika – Paul-Ehrlich-Institut)
| Bezeichnung | Zulassungs- /Genehmigungsinhaber | Indikation | Zulassungs- /Genehmigungsdatum | Erläuterungen |
|---|---|---|---|---|
| Abecma | Bristol-Myers Squibb Pharma EEIG | Multiples Myelom | 18.08.2021 | Seltene Erkrankung, CAR-T-Zell-Therapie |
| Beqvez (zuvor Durveqtix) | Pfizer Europe MA EEIG, Belgien | Hämophilie B | 24.07.2024 | Seltene Erkrankung |
| Breyanzi | Pharmaceuticals (Ireland) Limited | diffus großzelliges B-Zell-Lymphom (DLBCL); Hochgradiges B-Zell-Lymphom (HGBCL); primäres mediastinales großzelliges B-Zell-Lymphom (PMBCL); follikuläres Lymphom Grad 3B (FL3B) | 04.04.2022 | CAR-T-Zell-Therapie |
| Carvykti | Janssen-Cilag International N.V. | rezidiviertes oder refraktäres multiples Myelom | 25.05.2022 | Seltene Erkrankungen Erste CRISPR-Therapie |
| Casgevy | Vertex Pharmaceuticals (Ireland) Limited | Beta-Thalassämie und Sichelzellenanämie | 09.02.2024 | Seltene Erkrankungen Erste CRISPR-Therapie |
| Hemgenix | CSL Behring GmbH | Hämophilie B | 20.02.2023 | Seltene Erkrankung |
| Imlygic | Amgen Europe B.V. | Melanom | 16.12.2015 | onkolytisches Virus |
| Kymriah | Novartis Europharm Ltd., IRL | akute lymphatische B-Zell-Leukämie sowie diffus großzelliges B-Zell-Lymphom | 23.08.2018 | Seltene Erkrankungen, CAR-T-Zell-Therapie |
| Libmeldy | Orchard Therapeutics (Netherlands) B.V., NL | metachromatische Leukodystrophie | 17.12.2020 | Seltene Erkrankung |
| Luxturna | Novartis Europharm Limited | Netzhautdystrophie | 22.11.2018 | Seltene Erkrankung |
| Roctavian | BioMarin International Limited, Irland | Hämophilie A | 24.08.2022 | Seltene Erkrankung |
| Strimvelis | Fondazione Telethon ETS | Adenosin-Desaminase-Mangel (ADA-SCID) | 26.05.2016 | Seltene Erkrankung |
| Tecartus | Fondazione Telethon ETS | rezidiviertes oder refraktäres Mantelzelllymphom | 14.12.2020 | Seltene Erkrankung, CAR-T-Zell-Therapie |
| Upstaza | PTC Therapeutics International Limited, Irland | Aromatische L-Aminosäure-Decarboxylase (AADC)-Mangel | 18.07.2022 | Seltene Erkrankung |
| Yescarta | Kite Pharma EU B.V., NL | Hochgradiges B-Zell-Lymphom (HGBL); diffuses großzelliges B-Zell-Lymphom (DLBCL); primäres mediastinales großzelliges B-Zell-Lymphom (PMBCL); follikuläres Lymphom (FL) | 23.08.2018 | Seltene Erkrankungen. CAR-T-Zell-Therapie |
| Zolgensma | Novartis Europharm Limited, Dublin | Spinale Muskelatrophie (SMA) | 18.05.2020 | Seltene Erkrankung |
Quellen:
Gentherapeutika – Paul-Ehrlich-Institut
Gentherapie | Seltene Erkrankungen
Meldungen – Erstes Gentherapeutikum gegen Hämophilie A erhält Zulassung – Paul-Ehrlich-Institut
Chorea Huntington – Symptome, Diagnostik, Therapie | Gelbe Liste
Korrekturen an Chromosom 4: Gentherapien sollen Huntington-Krankheit verhindern
Programme & Pipeline | uniQure
Orphan Drug-Status im AMNOG-Verfahren erklärt | vfa
Die Gen-Reparateure – scinexx.de
Vergleich der regulatorischen Anforderungen an Gentherapeutika und genbasierte Impfstoffe